ScaffoldGraph Save
ScaffoldGraph is an open-source cheminformatics library, built using RDKit and NetworkX, for the generation and analysis of scaffold networks and scaffold trees.




![]()
![]()
⌬ ScaffoldGraph ⌬
ScaffoldGraph is an open-source cheminformatics library, built using RDKit and NetworkX, for the generation and analysis of scaffold networks and scaffold trees.
Features | Installation | Quick-start | Examples | Contributing | References | Citation
Features
-
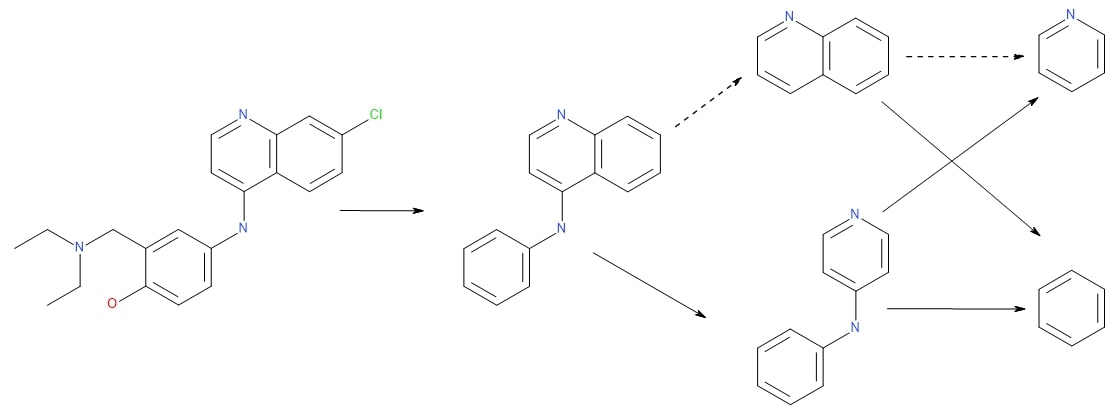
Scaffold Network generation (Varin, 2011)
- Explore scaffold-space through the iterative removal of available rings, generating all possible sub-scaffolds for a set of input molecules. The output is a directed acyclic graph of molecular scaffolds
-
HierS Network Generation (Wilkens, 2005)
- Explore scaffold-space through the iterative removal of available rings, generating all possible sub-scaffolds without dissecting fused ring-systems
-
Scaffold Tree generation (Schuffenhauer, 2007)
- Explore scaffold-space through the iterative removal of the least-characteristic ring from a molecular scaffold. The output is a tree of molecular scaffolds
-
Murcko Fragment generation (Bemis, 1996)
- Generate a set of murcko fragments for a molecule through the iterative removal of available rings.
-
Compound Set Enrichment (Varin, 2010, 2011)
- Identify active chemical series from primary screening data
Comparison to existing software
- Scaffold Network Generator (SNG) (Matlock 2013)
- Scaffold Hunter (SH) (Wetzel, 2009)
- Scaffold Tree Generator (STG) (SH CLI predecessor)
| SG | SNG | SH | STG | |
|---|---|---|---|---|
| Computes Scaffold Networks | X | X | - | - |
| Computes HierS Networks | X | - | - | - |
| Computes Scaffold Trees | X | X | X | X |
| Command Line Interface | X | X | - | X |
| Graphical Interface | - * |
- | X | - |
| Accessible Library | X | - | - | - |
| Results can be computed in parallel | X | X | - | - |
Benchmark for 150,000 molecules ** |
15m 25s | 27m 6s | - | - |
| Limit on input molecules | N/A *** |
10,000,000 | 200,000 **** |
10,000,000 |
* While ScaffoldGraph has no explicit GUI, it contains functions for interactive scaffoldgraph visualization.
** Tests performed on an Intel Core i7-6700 @ 3.4 GHz with 32GB of RAM, without parallel processing. I could not find
the code for STG and do not intend to search for it, SNG report that both itself and SH are both faster in the
benchmark test.
*** Limited by available memory
**** Graphical interface has an upper limit of 2,000 scaffolds
Installation
- ScaffoldGraph currently supports Python 3.6 and above.
Install with conda (recommended)
conda config --add channels conda-forge
conda install -c uclcheminformatics scaffoldgraph
Install with pip
# Basic installation.
pip install scaffoldgraph
# Install with ipycytoscape.
pip install scaffoldgraph[vis]
# Install with rdkit-pypi (Linux, MacOS).
pip install scaffoldgraph[rdkit]
# Install with all optional packages.
pip install scaffoldgraph[rdkit, vis]
Warning: rdkit cannot be installed with pip, so must be installed through other means
Update (17/06/21): rdkit can now be installed through the rdkit-pypi wheels for Linux and MacOS, and can be installed alongside ScaffoldGraph optionally (see above instructions).
Update (16/11/21): Jupyter lab users may also need to follow the extra installation instructions here / here when using the ipycytoscape visualisation utility.
Quick Start
CLI usage
The ScaffoldGraph CLI is almost analogous to SNG consisting of a two step process (Generate --> Aggregate).
ScaffoldGraph can be invoked from the command-line using the following command:
$ scaffoldgraph <command> <input-file> <options>
Where "command" is one of: tree, network, hiers, aggregate or select.
-
Generating Scaffold Networks/Trees
The first step of the process is to generate an intermediate scaffold graph. The generation commands are: network, hiers and tree
For example, if a user would like to generate a network from two files:
$ ls file_1.sdf file_2.sdfThey would first use the commands:
$ scaffoldgraph network file_1.sdf file_1.tmp $ scaffoldgraph network file_2.sdf file_2.tmpFurther options:
--max-rings, -m : ignore molecules with # rings > N (default: 10) --flatten-isotopes -i : remove specific isotopes --keep-largest-fragment -f : only process the largest disconnected fragment --discharge-and-deradicalize -d : remove charges and radicals from scaffolds -
Aggregating Scaffold Graphs
The second step of the process is aggregating the temporary files into a combined graph representation.
$ scaffoldgraph aggregate file_1.tmp file_2.tmp file.tsvThe final network is now available in 'file.tsv'. Output formats are explained below.
Further options:
--map-mols, -m <file> : generate a file mapping molecule IDs to scaffold IDs --map-annotations <file> : generate a file mapping scaffold IDs to annotations --sdf : write the output as an SDF file -
Selecting Subsets
ScaffoldGraph allows a user to select a subset of a scaffold network or tree using a molecule-based query, i.e. selecting only scaffolds for molecules of interest.
This command can only be performed on an aggregated graph (Not SDF).
$ scaffoldgraph select <graph input-file> <input molecules> <output-file> <options>Options:
<graph input-file> : A TSV graph constructed using the aggregate command <input molecules> : Input query file (SDF, SMILES) <output-file> : Write results to specified file --sdf : Write the output as an SDF file -
Input Formats
ScaffoldGraphs CLI utility supports input files in the SMILES and SDF formats. Other file formats can be converted using OpenBabel.
-
Smiles Format:
ScaffoldGraph expects a delimited file where the first column defines a SMILES string, followed by a molecule identifier. If an identifier is not specified the program will use a hash of the molecule as an identifier.
Example SMILES file:
CCN1CCc2c(C1)sc(NC(=O)Nc3ccc(Cl)cc3)c2C#N CHEMBL4116520 CC(N1CC(C1)Oc2ccc(Cl)cc2)C3=Nc4c(cnn4C5CCOCC5)C(=O)N3 CHEMBL3990718 CN(C\C=C\c1ccc(cc1)C(F)(F)F)Cc2coc3ccccc23 CHEMBL4116665 N=C1N(C(=Nc2ccccc12)c3ccccc3)c4ccc5OCOc5c4 CHEMBL4116261 ...-
SDF Format:
ScaffoldGraph expects an SDF file, where the molecule identifier is specified in the title line. If the title line is blank, then a hash of the molecule will be used as an identifier.
Note: selecting subsets of a graph will not be possible if a name is not supplied
-
-
Output Formats
-
TSV Format (default)
The generate commands (network, hiers, tree) produce an intermediate tsv containing 4 columns:
- Number of rings (hierarchy)
- Scaffold SMILES
- Sub-scaffold SMILES
- Molecule ID(s) (top-level scaffolds (Murcko))
The aggregate command produces a tsv containing 4 columns
- Scaffold ID
- Number of rings (hierarchy)
- Scaffold SMILES
- Sub-scaffold IDs
-
SDF Format
An SDF file can be produced by the aggregate and select commands. This SDF is formatted according to the SDF specification with added property fields:
- TITLE field = scaffold ID
- SUBSCAFFOLDS field = list of sub-scaffold IDs
- HIERARCHY field = number of rings
- SMILES field = scaffold canonical SMILES
-
Library usage
ScaffoldGraph makes it simple to construct a graph using the library API. The resultant graphs follow the same API as a NetworkX DiGraph.
Some example notebooks can be found in the 'examples' directory.
import scaffoldgraph as sg
# construct a scaffold network from an SDF file
network = sg.ScaffoldNetwork.from_sdf('my_sdf_file.sdf')
# construct a scaffold tree from a SMILES file
tree = sg.ScaffoldTree.from_smiles('my_smiles_file.smi')
# construct a scaffold tree from a pandas dataframe
import pandas as pd
df = pd.read_csv('activity_data.csv')
network = sg.ScaffoldTree.from_dataframe(
df, smiles_column='Smiles', name_column='MolID',
data_columns=['pIC50', 'MolWt'], progress=True,
)
Advanced Usage
-
Multi-processing
It is simple to construct a graph from multiple input source in parallel, using the concurrent.futures module and the sg.utils.aggregate function.
from concurrent.futures import ProcessPoolExecutor from functools import partial import scaffoldgraph as sg import os directory = './data' sdf_files = [f for f in os.listdir(directory) if f.endswith('.sdf')] func = partial(sg.ScaffoldNetwork.from_sdf, ring_cutoff=10) graphs = [] with ProcessPoolExecutor(max_workers=4) as executor: futures = executor.map(func, sdf_files) for future in futures: graphs.append(future) network = sg.utils.aggregate(graphs) -
Creating custom scaffold prioritisation rules
If required a user can define their own rules for prioritizing scaffolds during scaffold tree construction. Rules can be defined by subclassing one of four rule classes:
BaseScaffoldFilterRule, ScaffoldFilterRule, ScaffoldMinFilterRule or ScaffoldMaxFilterRule
When subclassing a name property must be defined and either a condition, get_property or filter function. Examples are shown below:
import scaffoldgraph as sg from scaffoldgraph.prioritization import * """ Scaffold filter rule (must implement name and condition) The filter will retain all scaffolds which return a True condition """ class CustomRule01(ScaffoldFilterRule): """Do not remove rings with >= 12 atoms if there are smaller rings to remove""" def condition(self, child, parent): removed_ring = child.rings[parent.removed_ring_idx] return removed_ring.size < 12 @property def name(self): return 'custom rule 01' """ Scaffold min/max filter rule (must implement name and get_property) The filter will retain all scaffolds with the min/max property value """ class CustomRule02(ScaffoldMinFilterRule): """Smaller rings are removed first""" def get_property(self, child, parent): return child.rings[parent.removed_ring_idx].size @property def name(self): return 'custom rule 02' """ Scaffold base filter rule (must implement name and filter) The filter method must return a list of filtered parent scaffolds This rule is used when a more complex rule is required, this example defines a tiebreaker rule. Only one scaffold must be left at the end of all filter rules in a rule set """ class CustomRule03(BaseScaffoldFilterRule): """Tie-breaker rule (alphabetical)""" def filter(self, child, parents): return [sorted(parents, key=lambda p: p.smiles)[0]] @property def name(self): return 'custom rule 03'Custom rules can subsequently be added to a rule set and supplied to the scaffold tree constructor:
ruleset = ScaffoldRuleSet(name='custom rules') ruleset.add_rule(CustomRule01()) ruleset.add_rule(CustomRule02()) ruleset.add_rule(CustomRule03()) graph = sg.ScaffoldTree.from_sdf('my_sdf_file.sdf', prioritization_rules=ruleset)
Contributing
Contributions to ScaffoldGraph will most likely fall into the following categories:
- Implementing a new Feature:
- New Features that fit into the scope of this package will be accepted. If you are unsure about the idea/design/implementation, feel free to post an issue.
- Fixing a Bug:
- Bug fixes are welcomed, please send a Pull Request each time a bug is encountered. When sending a Pull Request please provide a clear description of the encountered bug. If unsure feel free to post an issue
Please send Pull Requests to: http://github.com/UCLCheminformatics/ScaffoldGraph
Testing
ScaffoldGraphs testing is located under test/. Run all tests using:
$ python setup.py test
or run an individual test: pytest --no-cov tests/core
When contributing new features please include appropriate test files
Continuous Integration
ScaffoldGraph uses Travis CI for continuous integration
References
- Bemis, G. W. and Murcko, M. A. (1996). The properties of known drugs. 1. molecular frameworks. Journal of Medicinal Chemistry, 39(15), 2887–2893.
- Matlock, M., Zaretzki, J., Swamidass, J. S. (2013). Scaffold network generator: a tool for mining molecular structures. Bioinformatics, 29(20), 2655-2656
- Schuffenhauer, A., Ertl, P., Roggo, S., Wetzel, S., Koch, M. A., and Waldmann, H. (2007). The scaffold tree visualization of the scaffold universe by hierarchical scaffold classification. Journal of Chemical Information and Modeling, 47(1), 47–58. PMID: 17238248.
- Varin, T., Schuffenhauer, A., Ertl, P., and Renner, S. (2011). Mining for bioactive scaffolds with scaffold networks: Improved compound set enrichment from primary screening data. Journal of Chemical Information and Modeling, 51(7), 1528–1538.
- Varin, T., Gubler, H., Parker, C., Zhang, J., Raman, P., Ertl, P. and Schuffenhauer, A. (2010) Compound Set Enrichment: A Novel Approach to Analysis of Primary HTS Data. Journal of Chemical Information and Modeling, 50(12), 2067-2078.
- Wetzel, S., Klein, K., Renner, S., Rennerauh, D., Oprea, T. I., Mutzel, P., and Waldmann, H. (2009). Interactive exploration of chemical space with scaffold hunter. Nat Chem Biol, 1875(8), 581–583.
- Wilkens, J., Janes, J. and Su, A. (2005). HierS: Hierarchical Scaffold Clustering Using Topological Chemical Graphs. Journal of Medicinal Chemistry, 48(9), 3182-3193.
Citation
If you use this software in your own work please cite our paper, and the respective papers of the methods used.
@article{10.1093/bioinformatics/btaa219,
author = {Scott, Oliver B and Chan, A W Edith},
title = "{ScaffoldGraph: an open-source library for the generation and analysis of molecular scaffold networks and scaffold trees}",
journal = {Bioinformatics},
year = {2020},
month = {03},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa219},
url = {https://doi.org/10.1093/bioinformatics/btaa219},
note = {btaa219}
eprint = {https://academic.oup.com/bioinformatics/advance-article-pdf/doi/10.1093/bioinformatics/btaa219/32984904/btaa219.pdf},
}
