Mixcr Versions Save
MiXCR is an ultimate software platform for analysis of Next-Generation Sequencing (NGS) data for immune profiling.
v4.6.0
4 months ago🖇️ Combined Heavy+Light Somatic Hypermutation Trees from Single-Cell data
- A special step is added in
findShmTreesto combine heavy and light SHM trees utilizing information added to clonotypes bygroupClonescommand. Nodes in resulting tree will contain both light and heavy chains. If there is no connection to a clone from a companion chain, a reconstructed sequence will be added. - Behaviour can be disabled with
--dont-combine-tree-by-cellsoption to reconstruct separate heavy and light SHM trees - Added
exportShmSingleCellTreescommand that export one node per line. It there is several roots in a tree, data will be exported in a different columns. - Added
-subtreeIdto tree exports to differentiate part of trees from different chains -
exportShmTreesWithNodesandexportShmTreescommands will export subtrees with different chains at separate rows.
🚀 Other major upgrades
Changes in groupClones command
- Previous algorithm replaced with a new one that have better way of working with contamination, can detect multi-mappers (when one cell barcode marks two different cells) and can work with non-functional clones
- Some clones are now explicitly marked as contamination. This information is available as a separate label in
exportClonesingroupIdcolumn. Such clones can be filtered out from export by--filter-out-group-types contamination - More important algorithm performance metrics are added to the report
- Fix for behaviour leading to clones with
undefienedgroup being split by cell barcodes
New characteristics in SHM trees exports
-
-subtreeIdfor determination of different chains in the same tree -
-numberOfClonesInTree [forChain]Number of uniq clones in the SHM tree. -
-numberOfNodesWithClonesNumber of nodes with clones, i.e. nodes with different clone sequences. -
-totalReadsCountInTree [forChain]Total sum of read counts of clones in the SHM tree. -
-totalUniqueTagCountInTree (Molecule|Cell|Sample) [forChain]Total count of unique tags in the SHM tree with specified type. -
-chainsChain type of the tree -
-treeHeightHeight of the tree -
-vGene,-jGene,-vFamily,-jFamily- in previous version thous were exported only for nodes with clones -
-vBestIdentityPercent,-jBestIdentityPercent,-isOOFand-isProductivenow exported for reconstructed nodes too
New characteristics in clonotype export
-
-aaLengthand-allAALengthis available alongside-nLengthand-allNLength -
-aaMutationsRateis available alongside-nMutationsRate - Added optional arg
germlinein-nFeature,-aaFeature,-nLength,-aaLengthinexportClones,exportAlignmentsandexportCloneGroups. It allows to export a sequence of the germline instead of a sequence of the gene. - For all mutation exports (excluding
-mutationsDetailed) added optional filter by mutation type:... [(substitutions|indels|inserts|deletions)] - Added
-nMutationsCount,-aaMutationsCount,-allNMutationsCount,-allAAMutationsCountfor all relatable exports - For mutation exports in
exportShmTreesWithNodes(germline|mrca|parent)option is now optional. Will be export mutations fromgermlineby default - Added
--export-clone-groups-sort-chains-bymixin - Nucleotide mutations now could be exported for features that contain
VCDR3Part,DCDR3PartorJCDR3Part - Now
-nLength,-nMutationsCount,-nMutationsRatecan be calculated for multiple gene features (e.g.-nMutationsRate VRegionTrimmed,JRegionTrimmed) - Added
--export-clone-groups-sort-chains-bymixin with type of sorting of clones for determination of the primary and the secondary chains. It applies toexportCloneGroupscommand. By default, it'sAuto(by UMI if it's available, by Read otherwise; previous default value wasRead) - Added
--filter-out-group-typesmixin to filter-out clones having certain clone group assignment kind:found,undefinedorcontamination. It applies toexportClonescommand - Now
exportCloneGroupsby default will export groups in separate files forIG,TRAB,TRGDandmixed. This behaviour could be switched off by using--reset-export-clone-table-splittingor single--export-clone-groups-for-cell-type. In case of several--export-clone-groups-for-cell-typeevery cell type will be exported in separate file. - In case of
--export-clone-groups-for-cell-typeinexportCloneGroupsall mixed or unmatched groups will be filtered out. - Added read and Molecule fraction columns to single cell exportClones output.
🧬 Reference library upgrades
- Previous
TRADmeta-chain split intoTRAandTRDas it should be. Chain assignment for clonotypes based on J genes. - Rebuild allelic reference for human
IGH,TRB,TRAandTRDchains. Now allelic names correspond to the IUIS nomenclature. - Human
IGKVendcoordinates corrected. -
UTR5Begincoordinates added to the following mouse genes: IGKV23-1, IGKV20-101-2, IGKV14-130, IGKV8-28, TRGV2
📚 Preset updates
- The
milab-human-rna-tcr-umi-racepreset has been updated: now clones are assembled by default based on the CDR3, in line with the manufacturer's recommended read length. - The
flairr-seq-bcrpreset has been updated: now the preset sets species tohumanby default according to a built-in tag pattern with primer sequences. - The following presets have been added to cover Ivivoscribe assay panels:
invivoscribe-human-dna-trg-lymphotrack,invivoscribe-human-dna-trb-lymphotrack,invivoscribe-human-dna-igk-lymphotrack,invivoscribe-human-dna-ighv-leader-lymphotrack,invivoscribe-human-dna-igh-fr3-lymphotrack,invivoscribe-human-dna-igh-fr2-lymphotrack,invivoscribe-human-dna-igh-fr1-lymphotrack,invivoscribe-human-dna-igh-fr123-lymphotrack. - The following presets have been added for mouse Thermofisher assays:
thermofisher-mouse-rna-tcb-ampliseq-sr,thermofisher-mouse-dna-tcb-ampliseq-sr,thermofisher-mouse-rna-igh-ampliseq-sr,thermofisher-mouse-dna-igh-ampliseq-sr. - Preset for SMARTer Human scTCR a/b Profiling Kit:
takara-sc-human-rna-tcr-smarter - The
milab-human-rna-ig-umi-multiplexpreset has been updated: the pattern now trims fewer nucleotides, which facilitates CDR1 identification. The splits by V and J genes have been removed as redundant due to the full-length assembling feature.
🛠️ Minor improvements & fixes
- More strict
Combining treesstep infindShmTreescommand - Better calculation of indel mutations between nodes in process of building shm trees
- Increased percent of successful alignment-aided overlaps by removing unnecessary overlap region quality sum threshold
- Impossible export of germline sequence for
VJJunctioninshmTreesexports now produces an error - Parameter validation fix in
-nMutationsRate - Fix for
-nMutationsRateif region is not covered for the clone - Fix for the formal of
exportAlignmentsPrettybroken in the previous version - Fix for IllegalArgumentException in
exportAlignmentsPrettyfor cases where translation can't be performed - Fix error for
analyzeexecuted with-fand--output-not-used-readsat the same time - Resolutions of wildcards are excluded from calculation of
-nMutationsRatefor CDR3 inexportShmTreesWithNodes - Fix OutOfMemory exception in command
extendwith.vdjcainput - In
findShmTreesfilter for productive only clones now check for stop codons in all features, not only in CDR3 - Change default value for filter for productive clones in
findShmTreesto false (was true before) - Add option
--productive-onlytofindShmTrees - Fixed parsing of
--export-clone-groups-for-cell-typeparameter - Fixed usage of
slicecommand on clnx files that weren't ordered by id. - In
slicenow default behaviour is to keep original ids. Previous behaviour available with--reassign-idsoption - Fixed parsing of composite gene features with offsets like
--assemble-clonotypes-by [VDJRegion,CBegin(0,10)] - Fixed parent directory creation for output of
exportClonesOverlap - Fixed
exportAirrin case of a clone with CDR3 that don't have VCDR3Part and JCDR3Part - Optimize calculation of ranks in clone set. Speeds up export with tags and several other places.
- Added
clone_idcolumn inexportAirr - Fixed
exportClonesin case of splitting file bytag:...if there is a clone that have several tags of requested level - Fixed calculation of
-nMutationsCount,-nMutationsRate,-aaMutationsCountand-aaMutationsRate. Previously in some cases it was calculated on different region, from what was requested. - Added
CellBarcodesWithFoundGroupsforgroupClonesQC checks - New filter
--no-featureinexportAlignmentsPretty - Fixed reporting in
align, now coverage takes into account alignment-aided overlap
❗ Breaking changes
- Option
--build-from <path>was removed fromfindShmTreescommand
Deprecations of export options
-
-lengthOfnow is deprecated, use-nLengthinstead -
-allLengthOfnow is deprecated, use-allNLengthinstead -
-mutationRatenow is deprecated, use-nMutationsRateinstead
v4.5.0
7 months ago🚀 New features
Multi-chain clone assembly for single-cell data
Now MiXCR calculates Heavy-Light antibody and Alpha-Beta and Gamma-Delta TCR combined clones for single-cell data. Two new commands were introduced to enable this functionality:
-
groupClones: calculates multi-chain clones from assembled clonotypes and writes result in a binary format; -
exportCloneGroups: export information about combined clonotypes.
All single-cell presets now automatically produce combined multi-chain output in both binary and textual formats, see files with names matching *.clone.groups.tsv pattern in the output folder.
New characteristics in clonotype export
- Export biochemical properties of gene regions with
-biochemicalProperty <geneFeature> <property>or-baseBiochemicalProperties <geneFeature>export options. Available in export for alignments, clones and SHM tree nodes. Available properties:Hydrophobicity,Charge,Polarity,Volume,Strength,MjEnergy,Kf1,Kf2,Kf3,Kf4,Kf5,Kf6,Kf7,Kf8,Kf9,Kf10,Rim,Surface,Turn,Alpha,Beta,Core,Disorder,N2Strength,N2Hydrophobicity,N2Volume,N2Surface. - Export isotype with
-isotype [<(primary|subclass|auto)>] - Export
-mutationRate [<gene_feature>]inexportShmTreesWithNodes,exportClonesandexportCloneGroupscommand: number of mutations relative to corresponding germline divided by the target sequence size. ForexportClonesandexportCloneGroupsCDR3 is not included in calculation.
Support for wider set of input formats
- Support for
cramfiles as input foranalyzeandaligncommands. Optionally, a reference to the genome can be specified by--reference-for-cram - Fixed usage of BAM input for
analyzeandalign, if file contains both paired and single reads
Algorithm enhancements
- Global consensus assembly algorithm, applied in
assembleto collapse UMI/Cell groups into contigs, now have much better seed selection empirical step for multi-consensus assembly scenarios. This significantly increases sensitivity during assembly of secondary consensuses from the same group of sequences. - New constrain in low-quality reads mapping procedure preventing cross-cell read mapping.
📚 Preset updates
- Additional improvement of clone filters in
10x-sc-xcr-vdjpreset. - Tag pattern upgrade for
cellecta-human-rna-xcr-umi-drivermap-air. Now UMI includes a part of the C-gene primer to increase diversity, and R2 is also used for payload. - Assembling feature fix for
irepertoire-human-rna-xcr-repseq-pluspreset. Now{CDR2Begin:FR4End}. - New preset for BD full-length protocol with enhanced beads V2 featuring B384 whitelists:
bd-sc-xcr-rhapsody-full-length-enhanced-bead-v2. - New preset for Takara Bio SMART-Seq Mouse TCR (with UMIs):
takara-mouse-rna-tcr-umi-smarseq. - Presets for new Cellecta kits:
cellecta-human-dna-xcr-umi-drivermap-air,cellecta-human-rna-xcr-full-length-umi-drivermap-air,cellecta-mouse-rna-xcr-umi-drivermap-air. - Presets for iRepertoire RepSeq+ kits with UMI:
irepertoire-mouse-rna-xcr-repseq-plus-umi-pe,irepertoire-human-rna-xcr-repseq-plus-umi-se,irepertoire-human-rna-xcr-repseq-plus-umi-pe. -
isotypefield added toexportClonesfor presets supporting isotype identification. - Split by C-gene enabled in
thermofisher-human-rna-igh-oncomine-lrandcellecta-human-rna-xcr-umi-drivermap-airpresets to facilitate isotype separation. - Default consensus assembly parameters
maxNormalizedAlignmentPenaltyandaltSeedPenaltyToleranceare adjusted to increase sensitivity. - The
--split-by-sampleoption is now set totrueby default for allalignpresets, as well as all presets that inherit from it. This new default behavior applies unless it is directly overridden in the preset or with--dont-split-by-samplemix-in. -
exportAlignmentsnow reports UMI and/or Cell barcodes by default for presets with barcodes.
🛠️ Minor improvements & fixes
- Fixed possible crash with
--dry-runoption inanalyze - More informative help message that appears when using a deprecated preset and incorrectly suggests using
--assemble-contigs-byinstead of--assemble-clonotypes-by. - When split-by-tags is enabled,
exportCloneandexportShmTreesWithNodesnow output read count as the sum of reads for given tags selection, more complicated formula was used in previous versions -
exportAlignmentsby default now include the columntopChains.exportClonesfunction reportstopChainsfor single cell presets. - Fixed calculation of
geneFamilyNamefor genes likeIGHA*00(without the number before*symbol) - Better formatting in
listPresetscommand. Added grouping by vendor, labels and optional filtering - Validation of input types in
alignoranalyzeby given tag pattern
v4.4.2
9 months ago🚀 New features
- Two-fold
alignstep speedup for most of the protocol-specific presets (see the list below) - Import tags from sequence headers by parsing its content with regular expressions
- Highly optimized generic presets for amplicon/rna-seq sequence by long-read sequencers such as Pacific Biosciences and Oxford Nanopore:
generic-ont,generic-ont-with-umi,generic-pacbio,generic-pacbio-with-umi
🐞 Bug fixes
- Fixes crush when input contains quality scores > 70
- Fixes excessive memory consumption issue for long read data
- Fix for crush in
assemblewith UMI tags but with consensus assembler turned off
👷 Other minor adjustments
- Long-read J gene aligner optimization
- FLAIRR-seq preset optimized with new long-read-optimized aligner
- Quality trimming is disabled for long-read aligner
- Removed qc reports for clustered alignments and clones
- The following presets have been optimized by specifying a single reverse/direct alignment mode and now work faster:
takara-human-rna-bcr-umi-smartseq,takara-human-rna-bcr-umi-smarter,takara-human-rna-tcr-umi-smartseq,takara-human-rna-tcr-umi-smarter-v2,takara-human-rna-tcr-smarter,takara-mouse-rna-bcr-smarter,takara-mouse-rna-tcr-smarter,10x-sc-xcr-vdj,10x-sc-5gex,abhelix-human-rna-xcr,bd-human-sc-xcr-rhapsody-cdr3,bd-mouse-sc-xcr-rhapsody-cdr3,bd-sc-xcr-rhapsody-full-length,cellecta-human-rna-xcr-umi-drivermap-air,illumina-human-rna-trb-ampliseq-sr,illumina-human-rna-trb-ampliseq-plus,irepertoire-human-rna-xcr-repseq-sr,irepertoire-human-rna-xcr-repseq-lr,irepertoire-mouse-rna-xcr-repseq-sr,irepertoire-mouse-rna-xcr-repseq-lr,irepertoire-human-rna-xcr-repseq-plus,irepertoire-mouse-rna-xcr-repseq-plus,irepertoire-human-dna-xcr-repseq-sr,irepertoire-human-dna-xcr-repseq-lr,milab-human-rna-ig-umi-multiplex,milab-human-rna-tcr-umi-race,milab-human-rna-tcr-umi-multiplex,milab-human-dna-tcr-multiplex,milab-human-dna-xcr-7genes-multiplex,milab-mouse-rna-tcr-umi-race,neb-human-rna-xcr-umi-nebnext,qiagen-human-rna-tcr-umi-qiaseq
v4.4.0
10 months ago🚀 New features
Built-in alleles database
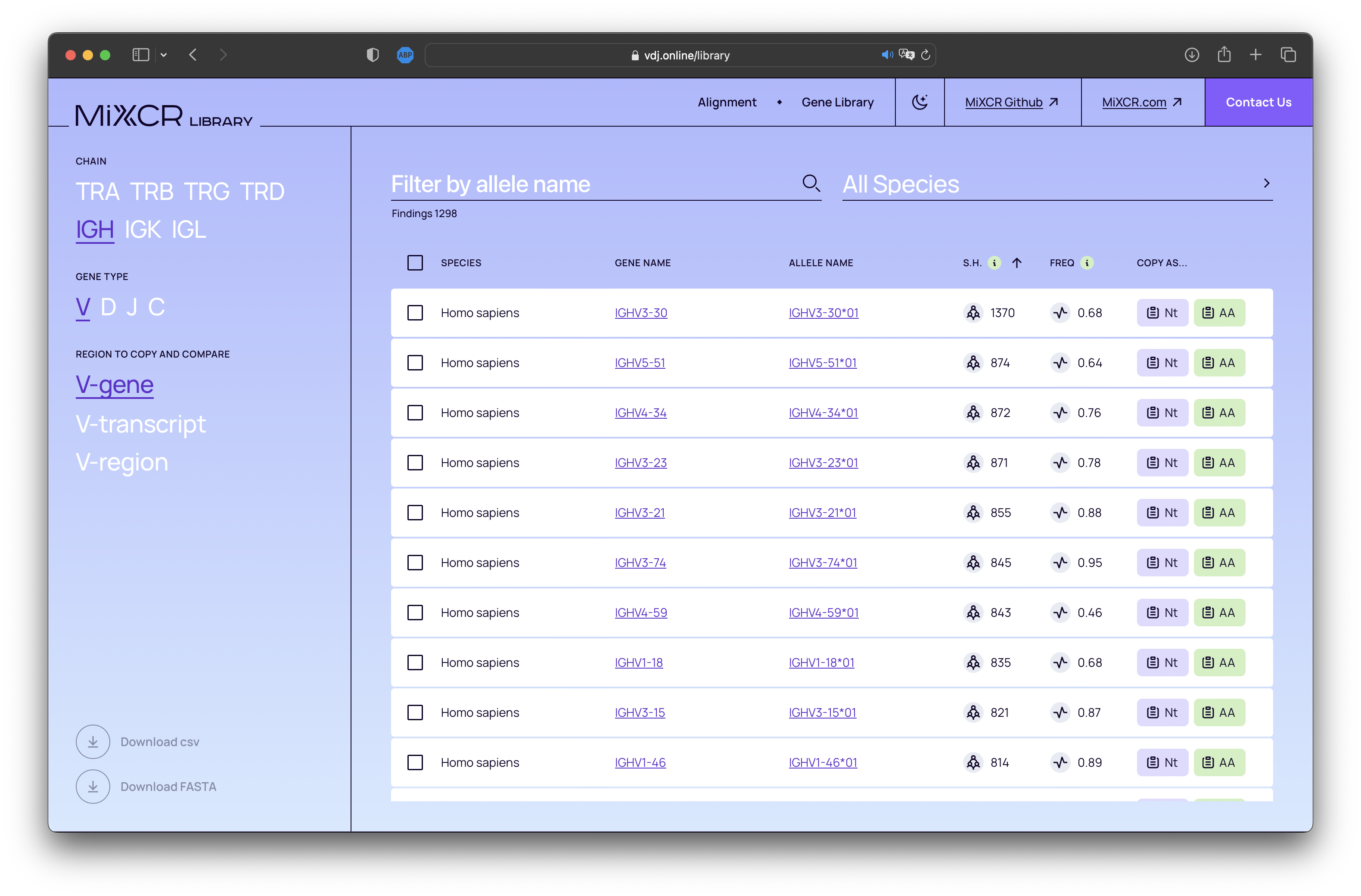
MiXCR features robust support for inferring donor-specific allelic variants of V and J genes from NGS data, using the findAlleles command. With this new release, we introduce a comprehensive built-in database of human alleles. Now, the findAlleles command will utilize known allele names from this integrated library. Feel free to explore our database at https://vdj.online/library.
New rigorous quality checks
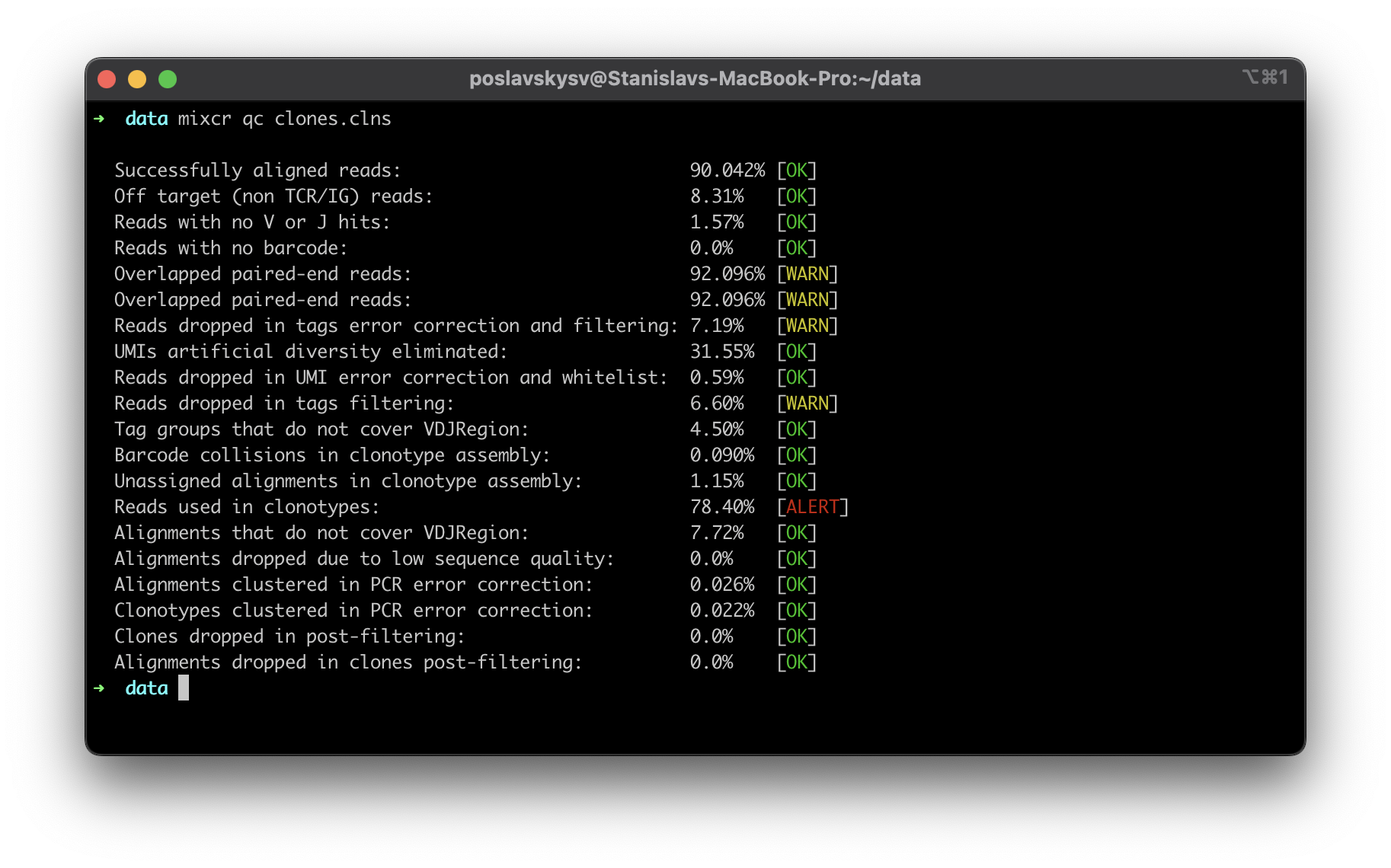
MiXCR now offers detailed insights into the quality of input data with its new quality control (QC) checks. A comprehensive list of checks provides complete information about the data and facilitates immediate feedback to the wet lab if any issues are detected
Convenient way to build custom libraries
Now one can build gene segment reference library for de-novo libraries or for chimeric model animals with just a single buildLibrary command. Check out our updated guide.
mixcr buildLibrary \
--v-genes-from-fasta v-genes.IGH.fasta \
--v-gene-feature VRegion \
--j-genes-from-fasta j-genes.IGH.fasta \
--d-genes-from-fasta d-genes.IGH.fasta \ # optional
--c-genes-from-fasta c-genes.IGH.fasta \ # optional
--chain IGH \
--species phocoena \
phocoena-IGH.json.gz
Comprehensive support of sample sheets
Now one can pass sample sheet directly to MiXCR analyze command as input. This way one can easily run MiXCR for arbitrary structure of input files, demultiplexed or not, with any type of multiplexing used:
mixcr analyze generic-sc-ht-vdj-amplicon --species hsa \
sample-sheet.csv \
output_prefix
🤩 New presets
-
Support of MiLaboratories Human 7 Genes DNA Multiplex:
milab-human-dna-xcr-7genes-multiplex -
Support of Parse Bio Evercode Whole Transcriptome presets:
parsebio-sc-3gex-evercode-wt-mini,parsebio-sc-3gex-evercode-wtandparsebio-sc-3gex-evercode-wt-mega -
Support of FLAIRR-Seq protocol via
flairr-seqpreset -
New generic single cell presets:
-
Low throughput (e.g. micro-wells) amplicon-based single cell:
- No UMIs:
generic-sc-lt-vdj-amplicon - With UMIs:
generic-sc-lt-vdj-amplicon-umi
- No UMIs:
-
Low throughput (e.g. micro-wells) single cell with fragmentation (RNA-Seq):
- No UMIs:
generic-sc-lt-vdj-fragmented - With UMIs:
generic-sc-lt-vdj-fragmented-umi
- No UMIs:
-
High throughput (e.g. droplets) amplicon-based single cell:
- No UMIs:
generic-sc-ht-vdj-amplicon - With UMIs:
generic-sc-ht-vdj-amplicon-umi
- No UMIs:
-
High throughput (e.g. droplets) single cell with fragmentation (RNA-Seq):
- No UMIs:
generic-sc-ht-vdj-fragmented - With UMIs:
generic-sc-ht-vdj-fragmented-umi
- No UMIs:
-
Reconstructing VDJ from generic gene expression data:
- No UMIs:
generic-sc-gex - With UMIs:
generic-sc-gex-umi
- No UMIs:
-
-
New Biomed2 primer sets:
biomed2-human-rna-igkl,biomed2-human-rna-trbdg.
💪 Major changes
-
Improved aligner parameters for all protocols. We spent in total more than 100,000 CPU/hours running optimization. As a result alignment rate is better for most of the protocols, especially in the case of average data quality.
-
Adds new
minSequenceCountparameter for k-mer filter, allowing construction of more flexible filtering pipelines with better fallback behaviour for under-sequenced libraries. -
Now full sample sheet with input file names can be provided as an input to the pipeline.
-
Sample sheets provided both with
--sample-sheetmixin and as a pipeline input, will be fuzzy matched against the data, allowing for one substitutions in unambiguous cases. This behaviour can be turned off by using--sample-sheet-strictmixin instead, or by adding a--strict-sample-sheet-matchingoption if full sample sheet input is used as pipeline input. -
New commands:
mixcr qc,mixcr buildLibrary) ,mixcr mergeLibrary,mixcr debugLibrary) -
Various major improvements to sequencing and PCR error correction algorithms for tags and clonotypes:
- tag refinement now uses average quality in statistical inference; this is the correct approach from the mathematical point of view, and it slightly increases performance judging by better consensus assembly downstream
- statistical inference in PCR error correction redone from scratch, now it takes into account aggregated quality scores of clonotypes, which makes the procedure automatically adapt to low quality samples and better perform in many marginal cases in both UMI and non-UMI protocols
- better algorithm for quality score aggregation in clonotype assembly
- better algorithm for quality score aggregation in consensus assembly
-
Mechanism to apply different tag transformations on the
alignstep. Transformations include mappings, string and sequence manipulations and various arithmetic operations. This feature allows to fit single-cell scenarios where multiple well-known barcodes marks the same cell, allows to convert sequence barcodes to textual representation to adopt different barcode naming schemas used in some protocols, convert multiple barcodes to single cell id. Feature is currently used in presets for analysis of data from Parse Bioscience and BD Rhapsody single-cell platforms. -
Special mechanism to allow for
NaNvalues in metrics in group filters (used inminSequenceCountparameter in k-mer filter, see below). -
Added fallback behaviour for under-sequenced libraries
🐞 Bug fixes
- Fix for naming of intermediate files and reports produced by
analyzeif target folder is specified - Tag pattern now is also searched in reverse strand for single-ended input with
--tag-parse-unstranded - fix for value in report line
Reads dropped due to low quality, percent of total report string - Fixed bug not allowing to parse more than two reads with tag pattern
- Fixed bug when
--chainsis used withexportClonesOverlap - Fixed for
export...- tag quality field added back to export columns
👷 Minor fixes and improvements
- Added gene feature coverage in alignment report
- On Linux platforms default calculation of -Xmx now based on "available" memory (previously "free" was used)
- New gene aligner parameter
edgeRealignmentMinScoreOverridefor more sensitive alignments for short paired-end reads - Report values downstream
alignnow calculate percents relative to the number of reads in the sample rather than the total number of reads in multi-sample analysis - Options helping with advanced analysis of data quality and consensus assembly process added
to
assemble(--consensus-alignments,--consensus-state-stat,--downsample-consensus-state-stat) andanalyze(--output-consensus-alignments,--output-consensus-state-stat,--downsample-consensus-state-stat) - Better tag pattern search projection representation in reports
-
findAllelesnow recalculate functionality of de novo found alleles - Better algorithm to calculating checksum of VDJC library
- Additional report string "Aligned reads processed" in
assemblereport - Added options
--by-featureand--by-genetosortClones - Added options
-rankByReadsand-rankByTag <(Molecule|Cell|Sample)>toexportClonesandexportShmTreesWithNodes.txt - Export
readIdsinexportAlignmentsby default - Added recalculation functionality for de-novo found alleles in
findAlleles - Add info about CDR3 in generated hash for de-novo alleles
- Remove de-novo alleles that are actually the same
-
findAlleleswill remove not used genes from the library (genes that not represented in given donor) - Make
--chainsoptional indownsamplingcommand and allow multiple input - Write empty file on
exportClonesif file doesn't contain any clones - Better exception messages on incorrect inputs for export commands
- In
exportCloneswriteno_d_geneif requestedVDJunction,DCDR3PartorDJJunctionin absence of D hit - Columns in
exportReportsTablenow covers most of significant statistics from reports
🐬 Docker image changes
-
Custom entry-point of the image removed, and now is set to
/bin/bash. Now one needs to specifymixcrcommand at the beginning of argument list:Old:
docker run ghcr.io/milaboratory/mixcr/mixcr analyze ...New:
docker run ghcr.io/milaboratory/mixcr/mixcr mixcr analyze ... -
New image is based on Amazon Corretto which in turn is based on Amazon Linux 2. If customization is required for the image, one now need to use
yumpackage manager instead ofapt/apt-get.With old image:
FROM ghcr.io/milaboratory/mixcr/mixcr:4.3.2 # ... RUN apt-get install -y wget # ...With new image:
FROM ghcr.io/milaboratory/mixcr/mixcr:4.4.0 # ... RUN yum install -y wget # ...see official docs for more detais.
-
Better compatibility of official docker image with Nextflow
‼️ Breaking changes
-
Parameter
clusteringFilter.specificMutationProbabilityremoved fromassembleaction. Three new parameters are introduced instead:-
clusteringFilter.correctionPower- this parameter determines how thorough the procedure should eliminate erroneous variants. Smaller value leaves less erroneous variants at the cost of accidentally correcting true variants. This value approximates the fraction of erroneous variants the algorithm will miss (type II errors). -
clusteringFilter.backgroundSubstitutionRate- expected rate of substitutions happening before the sequencing. -
clusteringFilter.backgroundIndelRate- expected rate of indels happening before the sequencing.
-
-
Majority of presets underwent name revisions (legacy names remain functional, though accompanied by deprecation warnings). See the full list of renames here.
v4.3.2
1 year ago🐞 This update addresses a significant issue that first appeared in version 4.3.0, which caused incorrect column names for FR4 nucleotide and amino acid sequences in export tables (e.g. nSeqJGeneWithoutCDR3Part instead of nSeqFR4).
Minor improvements
-
findAllelesnow works much faster for extremely diverse samples
Other bug fixes
- fixed inconsistency in reports and behaviour for
assemblewhenbadQualityThreshold=0 - fixes X axes label for k-mer filters in tags filtering QC plots
- adds threshold lines for tags filtering QC plots for composite operators (like operators with
cumtopfallbacks) - fixes NPE crash for chain usage plots if chimeric sequences present in the stats
v4.3.1
1 year agoMinor improvements
- added
-isOOF <gene_feature>column to export - added
-hasStops <gene_feature>column to export - added
-isProductive <gene_feature>column to export - improvements of report and alleles description table for
findAllelescommand - removing of unused genes from result library in
findAllelescommand -
findAllelesnow more resilient to case when most allele variants of donor differ from*00alleles in a library
Bug fixes
- fixed AssertionError in
findAllelescommand with--output-templateargument - fixed wrong behaviour with
inferMinRecordsPerConsensus == trueand cell level assembly - fixed
minRecordsPerConsensusinference mechanism for new filtering features introduced in previous version (4.3.0)
v4.3.0
1 year agoKey changes
- Improved Otsu's method with less stringency for automated histogram thresholding for barcoded data. It allows to recover more "good" UMI groups. The old filter was replaced by new one in all presets for airr-seq and single cell V(D)J protocols that utilize UMI: Cellecta, Milaboratories, NEB, Qiagen, Takara, 10x Genomics, BD, Singleron.
- New group filter operators allowing to mix thresholds form multiple operators, taking lowest or highest value and applying it. This allows to create more universal filtering strategies, robust to edge cases like undersequencing of barcodes.
- Added default fallback threshold for UMI filtering: if automated UMI thresholding leaves less than 85% of reads, then MiXCR will preserve UMIs to always keep minimum 85% of reads.
Presets
- New preset for Seq-Well VDJ data
- New presets for NEBNext® Immune Sequencing Kit TCR and BCR profiling for data with both TCR and BCR.
- Improved Takara human TCR and BCR presets
Reference Library
- New IGHV genes added to human reference: IGHV3-30-3, IGHV4-30-4, IGHV1-69-2, IGHV2-70D, IGHV3-30-5
- IGHV1-69D renamed to IGHV1-69
Minor improvements
- Threshold rounding in cumtop and top-n filters
- Support of sequence-end token (
$) in tag pattern matching algorithm - Added
discardAmbiguousNucleotideCallsparameters for contig assembly - Added field
-cellIdin commandsexportClonesandexportAlignments - Added fields
cell_id,umi_countandconsensus_counttoexportAirrcommand - Better text descriptions in align and assemble reports
-
exportAirrcommand now split clones by cells if there is cell barcodes in the data - Replace
analyzeoptions--not-aligned-..and--not-parsed-..with one option--output-not-used-reads - Fix comma-separated chains input in postanalysis
--chainsoption - Split column with tagValue (like
tagValueCELL) to two columns:tagValue<tag_name>andtagQuality<tag_name> - Support of system proxy settings for license
-
#character now can be used to separate groupName from group matcher in file expansion mechanist (additionally to:), allowing multi-sample analysis on Windows - Fixed usage of composite features for
--assemble-contigs-by - Removed some restrictions for possible combinations of gene features used in analysis and export
- Fixed behaviour of empirical alignment assignment in
assembleif--write-allwas used inalign
v4.2.0
1 year agoBuilt-in support for new protocols
-
BD Rpahsody full-length protocol
-
Smart-Seq2 single cell RNA-Seq protocol
-
Oxford Nanopore long-read technology
Sample barcodes
Complete support of sample barcodes that may be picked up from all possible sources:
- from names of input files;
- from index I1/I2 FASTQ files;
- from sequence header lines;
- from inside the tag pattern.
Now one can analyze multiple patient samples at once. Along with a powerful file name expansion functionality, one can process any kind of sequencing protocol with any custom combination of sample, cell and UMI barcoding.
Processing of multiple samples can be done in two principal modes in respect to sample barcodes: (1) data can be split by samples right on the align stage and processed separately, or (2) all samples can be processed as a single set of sequences and separated only on the very last exportClones step, both approaches have their pros and cons allowing to use the best strategy given the experimental setup and study goals.
New robust filters for single cell and molecular barcoded data
For 10x Genomics and other fragmented protocols, a new powerful k-mer based filtering algorithm is now used to eliminate cross-cell contamination coming from plasmatic cells.
For UMI filtering, a new algorithm from the paper by J. Barron (2020) allows for better automated histogram thresholding in barcoded data filtering.
List of all changes
Sample barcodes
- support for more than two
fastqfiles as input (I1andI2reads support) - multiple possible sources of data for sample resolution:
- sequences extracted with tag pattern (including those coming from
I1andI2reads) - samples can be based on specific pattern variant (with multi-variant patterns, separated by
||, allows to easily adopt MiGEC-style-like sample files) - parts of file names (extracted using file name expansion mechanism)
- sequences extracted with tag pattern (including those coming from
- flexible sample table matching criteria
- matching multiple tags
- matching variant id from multi-variant tag patterns
- special
--sample-tablemixin option allowing for flexible sample table definition in a tab-delimited table form - special
--infer-sample-tablemixin option to infer sample table for sample tags from file name expansion - special generic presets for multiplexed data analysis scenarios (e.g.
generic-tcr-amplicon-separate-samples-umi) -
aligncommand now optionally allows to split output alignments by sample into separatevdjcafiles -
exportClonescommand now supports splitting the output into multiple files by sample -
analyzecommand supports new splitting behaviour of thealigncommand, separately running all the analysis steps for all the output files (if splitting is enabled)
Filters and error correction
- preset for 10X VDJ BCR enhanced with k-mer-based filter to eliminate rare cross-cell contamination from plasmatic cells
- new advanced thresholding algorithm from the paper by J. Barron (2020) allows for better automated histogram thresholding in barcoded data filtering
- rework of clustering step aimed at PCR / reverse-transcription error correction in
assemble, now it correctly handles any possible tag combination (sample, cell or molecule) - new feature to add histogram preprocessing steps in automated thresholding
Quality trimming
- turn on default quality trimming (
trimmingQualityThresholdchanged from0to10), this setting showed better performance in many real world use-cases
Reference library
- reference V/D/J/C gene library upgrade to repseqio v2.1 (see changelog)
New commands
- added command
exportReportsTablethat prints file in tabular format with report data from commands that were run
Other
- optimized aligner parameters for long-read data
- fixed system temp folder detection behaviour, now mixcr respects
TMPDIRenvironment variable - rework of preset-mixin logic, now external presets (like those starting from
local:...) are packed into the output*.vdjcafile onalignstep, the same applies to all externally linked information, like tag whitelists and sample lists. This behaviour facilitates better analysis reproducibility and more transparent parameter logistics. - new mixin options to adjust tag refinement whitelists with
analyze:--set-whitelistand--reset-whitelist - removed
refineTagsAndSortoptions-wand--whitelist; corresponding deprecation error message printed if used - new grouping feature for
exportClones, allowing to normalize values for-readFractionand-uniqueTagFraction ...columns to totals for certain compartments instead of normalizing to the whole dataset. This feature allows to output e.g. fractions of reads inside the cell. - new mixin options
--add-export-clone-table-splitting,--reset-export-clone-table-splitting,--add-export-clone-groupingand--reset-export-clone-grouping - improved sensitivity of
findAllelescommand - add tags info in
exportAlignmentsPrettyandexportClonesPretty - add
--chainsfilter forexportShmTrees,exportShmTreesWithNodes,exportShmTreesNewickandexportPlots shmTreescommands - fixed old bug #353, now all aligners favor leftmost J gene in situations where multiple genes can ve found in the sequence (i.e. mis-spliced mRNA)
- fixes exception in
alignhappening for not-parsed sequences withwriteFailedAlignments=true - new filter and parameter added in
assemblePartial; parameter name isminimalNOverlapShare, it controls minimal relative part of N region that must be covered by the overlap to conclude that two reads are from the same V(D)J rearrangement - default paired-end overlap parameters changed to slightly more relaxed version
- better criteria for alignments to be accepted for the
assemblePartialprocedure - fixed NPE in
assemblePartialexecuted for the data without C-gene alignment settings - fixed rare exception in
exportAirrcommand - by default exports show messages like 'region_not_covered' for data that can't be extracted (requesting
-nFeaturefor not covered region or not existed tag). Option--not-covered-as-emptywill save previous behaviour - info about genes with enough data to find allele was added into report of
findAllelesand description of alleles - fixed error message appearing when analysis parameter already assigned to
nullis overridden bynullusing the-O...option - fixed wrong reporting of number of trimmed letters from the right side of R1 and R2 sequence
- fixed error message about repeated generic mixin overrides
- fixed error of
exportCloneswith some arguments - fixes for report indention artefacts
- fixed bug when chains filter set to
ALLinexportAlignmentswas preventing not-aligned records to be exported - fixed runtime exception in
assemblerising in analysis of data with CELL barcodes but without UMIs, with turned off consensus assembly - fixed bug leading to incorrect mixin option ordering during it's application to parameters bundle
- minor change to the contigAssembly filtering parametrization
- added mix-in
--export-productive-clones-only - warning message about automatically set
-Xmx..JVM option inmixcrscript - safer automatic value for
-Xms.. - fix: added
speciesflag to 10x, nanopore and smart-seq2 presets
v4.1.2
1 year agoMajor changes
- Command
findShmTreesnow can build trees from inputs with different tags - Added
--impute-germline-on-exportand--dont-impute-germline-on-exporttoexportAlignmentsandexportClonescommands
Minor improvements
- Now, instead of specifying separately multiple tags of the same type (i.e. CELL1+CELL2+CELL3) in filters, one can use
convenient aliases (like
allTags:Cell,allTags:Molecule). This also facilitates creation of a more generic base presets implementing common single-cell and UMI filtering strategies. - Several command line interface improvements
- Migration from
<tag_name>to<tag_type>semantics in export columns and--split-by-tagoptions
Fixes
- fixes bug with
saveOriginalReads=trueonalignleading to errors down the pipeline -
analyzenow correctly terminates on first error - correct progress reporting in
alignwith multiple input files provided by file name expansion mechanism - fix
--only-observedbehaviour inexportShmTreesWithNodes - fix missing tile in heatmap
- fix some cases of usage of
-O...
Presets
- Fixed issue with mouse presets from MiLaboratories
- Fixed presets with whitelists
- Fixed missing material type and species in several presets
- Added template switch region trimming for RACE protocols
- Added presets for
- Thermo Fisher Oncomine kits
- ParseBio single-cell protocols
- iRepertoire kits
- Preset for protocol described in Vergani et al. (2017)
- Cellecta AIR kit